Inflammasomes Mechanism of Assembly Regulation and Signaling Broz and Dixit Nat Imm Review
Introduction
A variety of pathogenic viruses can cause astringent diseases and are threats to human being health, such as hepatitis C virus (HCV), human being immunodeficiency virus-i (HIV-1), influenza A virus (IAV), and Zika virus (ZIKV). To eradicate invading viruses chop-chop and efficiently, host has evolved highly conserved sensors, called pattern recognition receptors (PRRs) (one), that recognize viral infections and, subsequently, trigger antiviral immune responses. PRRs, which include Cost-like receptors (TLRs), retinoic acid-inducible gene-I (RIG-I) like receptors (RLRs), and DNA sensors such equally cyclic GMP-AMP synthase (cGAS), sense different pathogen-associated molecular patterns (PAMPs), and damage-associated molecular patterns (DAMPs) derived from invading viruses. Upon engagement with their cognate ligands, PRRs can induce the activation of 2 unlike transcription factor-mediated pathways, IRF3 and NF-κB. IRF3 mediates the secretion of type I interferons (IFNs), which lead to the activation of the JAK-STAT pathway and the expression of interferon-stimulated genes (ISGs) (2). NF-κB initiates both the production of proinflammatory factors, such equally tumor necrosis factor (TNF)-α and interleukin (IL)-6, too as the initiation of inflammasome priming phase (come across below).
Some PRRs, such equally NACHT, LRR, and PYD domains-containing protein 1 (NLRP1), NLRP3, NLR family CARD domain-containing protein iv (NLRC4), and absent in melanoma 2 (AIM2), recruit apoptosis-associated speck-like protein (ASC) and caspase-one to course the inflammasome—a multimeric platform of proteins that initiates inflammation as well equally some forms of cell expiry (iii). Among all the inflammasomes discovered, the NLRP3 inflammasome is the virtually extensively studied and it plays an important office in both inflammation and antiviral responses. However, the mechanisms of the NLRP3 inflammasome activation are still complicated and remain controversial. In this review, we will focus on the contempo research advances made in terms of NLRP3 inflammasome activation during a viral infection and the immune evasion mechanisms of viruses that target the NLRP3 inflammasome.
The Activation Of The NLRP3 Inflammasome
The roles of the NLRP3 inflammasome are vital in the host antiviral immune responses. Several viruses, such every bit IAV and West Nile virus (WNV), tend to induce an appropriate and early phase activation of the NLRP3 inflammasome. As a upshot, activation of the NLRP3 inflammasome inhibits viral replication and reduces mortality in mouse models (4, 5). The NLRP3 inflammasome can be activated past sensing viral components too as cytosolic danger signals, such equally mitochondria injury, poly peptide aggregates, and aberrant ion concentrations, all of which can exist caused by a viral infection.
NRLP3 inflammasome activation requires two steps (Figure 1). The offset step, known as the priming step, is induced past PRRs or TNFR activation. This leads to the activation of NF-κB and promotes the expression of NLRP3, pro-IL-1β, and pro-IL-18. Additionally, IFNAR also activates the priming stage of NLRP3 inflammasome activation (6). The second step, also called the activation pace, is triggered past a range of stimuli that emerge during infections, tissue impairment, or metabolic imbalances. Such stimuli include ATP, pore-forming toxins, crystalline substances, nucleic acids, and invading pathogens (7). NLRP3 recruits ASC through its N-last pyrin domain (PYD) past homophilic interactions, resulting in the formation of ASC prion-like oligomerizes (8). The NAIP, CIITA, HET-E and TP1 (NACHT) domain in the middle of the NLRP3 possesses dNTPase action and mediates downstream oligomerization (ix). The C-terminal leucine-rich echo domain (LRR) associates with HSP90, SGT1, and PML and is considered to be responsible for the regulation of NLRP3 inflammasome activeness (10, 11).
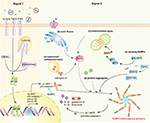
Figure 1. NLRP3 inflammasome activation during viral infections. Activation of the NLRP3 inflammasome requires two signals. Signal ane (priming bespeak): the activation of PRRs, TNFR, or IFNR induces NF-κB activation, triggers the transcription of NLRP3, pro-caspase-1, pro-IL-1β, and pro-IL-18. Signal 2 (activation signal): multiple DAMPs and PAMPs induce NLRP3 inflammasome assembly and activation. DAMPs include (a) lysosomal or endosomal injury, (b) aberrant ionic fluxes, (c) mitochondrial injury, and (d) poly peptide aggregates. (east) With the assist of DAI/ZBP1, DHX33, OAS, or DDX19A, NLRP3 is activated by sensing viral proteins and RNA. NLRP3 inflammasome activation leads to the auto-cleavage of pro-caspase-i. Caspase-1 then mediates the proteolytic procedure of pro-IL-1β, pro-IL-eighteen, and gasdermin D (GSDMD).
Once assembled, the NLRP3 inflammasome triggers the auto-cleavage of pro-caspase-i (12, 13). Equally an effect factor, caspase-1 mediates the proteolytic processing of pro-IL-1β, pro-IL-xviii, and the propyroptotic factor gasdermin D (GSDMD) (14). GSDMD forms pores in the membrane of infected cells, facilitating the secretion of IL-1β/IL-18 and inducing the inflammation-associated cell death known every bit pyroptosis (fifteen). The secretion of IL-1β subsequently recruits neutrophils to the inflammatory site to help in the elimination of invading viruses (16). Moreover, both IL-1β and IL-18 are responsible for the subsequent induction of the adaptive allowed response (17, 18). Accordingly, optimal activation of the NLRP3 inflammasome facilitates the institution of a host antiviral status.
However, aberrant NLRP3 inflammasome activation tin likewise lead to severe pathological injury. In an IAV infection model, juvenile mice had sustained elevated levels of blazon I IFNs and persistent NLRP3 inflammasome activation, suffering from severe lung injury independent of viral titer (xix). In improver, HIV-1 infected microglia are shown to cause NLRP3-associated neuroinflammation (20). HCV infection promotes chronic intrahepatic inflammation and liver injury mediated by the NLRP3 inflammasome (21). The recruitment of excessive inflammatory cells and pyroptosis-mediated cell impairment take role in the immunopathology progresses (22, 23).
Priming Footstep Of NLRP3 Inflammasome Activation In Viral Infection
In the rest state, cellular NLRP3 level is low enough to avoid aberrant inflammasome assembly and activation. Viral infection initiates NF-κB signaling through the activation of PRRs-dependent pathways (24, 25). RLRs, as cytosolic RNA sensors, discover viral RNA, such as VSV and IAV (26, 27). TLR3, TLR7, and TLR9 participate in the sensing of IAV, HCV, and adenovirus type 5 (Ad5) (21, 27, 28). Respiratory syncytial virus (RSV), IAV and human parainfluenza virus (HPIV) activate TLR2 or TLR4 in macrophages (29–31). Canker simplex virus type ane (HSV-one) infection could be detected by cGAS, a major cytosolic Dna sensor (32). Research indicates that HIV can prime number NLRP3 inflammasome transcription in monocyte-derived macrophages (33). Moreover, PRRs induced IFN-β and TNF-α that could, in plow, activate NF-κB and provide the cascade amplification necessary for NLRP3 inflammasome activation. This response enables the host to defend effectively against viral infections.
Viral Infection Triggered The Activation Step Of NLRP3 Inflammasome
The NLRP3 inflammasome tin can be activated past both viral components, including RNA and proteins (PAMPs), and danger signals (DAMPs). Although it does not directly interact with viral structures, the NLRP3 inflammasome is nonetheless sensitive to invading viruses and cytosolic danger signals, indicating its complicated mechanisms of sensing invading pathogens.
PAMPs
NLRP3 can sense some PAMPs with the aid of other receptors. In chief homo monocyte-derived macrophages, the DExD/H-box RNA helicase family member, DHX33, senses reoviral genomic RNA, interacts with NLRP3 to form the inflammasome complex, and leads to the secretion of IL-1β (34). two′,5′-oligoadenylate (two–5A) synthetase (OAS) recognizes dsRNA from some viruses, such as IAV and VSV, and promotes the cleavage thereof by endoribonuclease RNase L; the cleaved nucleic acids then be detected by DHX33 (35). Furthermore, DDX19A, some other DExD/H-box RNA helicase family member, senses porcine reproductive and respiration syndrome virus (PRRSV) and promotes NLRP3 inflammasome activation (36).
With the help of DAI/ZBP1, NLRP3 recognizes viral proteins and promotes inflammasome assembly (37). DAI/ZBP1 interacts with the IAV nucleoprotein (NP) and polymerase subunit PB1 after infection. DAI/ZBP1 after interacts with RIP3 through their shared domain homotypic interaction motif (RHIM), to activate the NLRP3 inflammasome via the RIP1-RIP3-caspase-8 pathway (37–39). It is evident that the viral protein sensor DAI/ZBP1 is critical to the induction of NLRP3 inflammasome-mediated apoptotic and necroptotic cell death since DAI/ZBP1 deficient mouse were protected from mortality during IAV infection. Even so, viral RNA or proteins alone were not sufficient to induce DAI/ZBP1-mediated prison cell decease during IAV infection (xl). Instead, during viral replication, DAI/ZBP1 senses the viral ribonucleoprotein (vRNP), containing IAV RNA, NP, and PB1, and later on initiates programmed cell death (40).
DAMPs
To date, no ligand that binds directly to NLRP3 has been found. Accordingly, the NLRP3 inflammasome is usually associated with sensing cytosolic danger signals referred to equally DAMPs. Not only intact viruses, such as IAV, SeV, HSV, and adenovirus (AdV), but also viral components, including internalized or genomic DNA, dsRNA, ssRNA, and fifty-fifty poly(I:C), could straight actuate the NLRP3 inflammasome and induce IL-1β secretion in macrophages (5, 28, 41–43). During infection, viruses cause a series of changes in cellular status of their host cells, including lysosomal maturation, aberrant ion concentrations, mitochondria damage, and the accumulation of misfolded protein aggregates, all of which are recognized equally danger signals by the host and lead to the activation of the NLRP3 inflammasome.
The maturation and acidification of lysosomes lead to the leaking of catalytically active cathepsin B, and the subsequent generation of reactive oxygen species (ROS), which, in turn, activates the NLRP3 inflammasome (5, 44). AdV blazon 5 induces the disruption of endosomal membranes and the release of cathepsin B, thereby activating NLRP3 (28, 45). This activation is required for the lysosomal localization and membrane penetration ability of AdV, since the temperature-sensitive mutant of Ad5 cannot induce the activation of NLRP3. ROS are also required for NLRP3 inflammasome activation since lower levels of IL-1β were observed in the presence of NADPH oxidase inhibitors or the oxygen scavenger N-acetylcysteine. IAV infection, or RNA species, activates the NLRP3 inflammasome by inducing lysosomal acidification (5).
An appropriate ionic concentration is crucial to maintain cellular homeostasis inside host cells. Nonetheless, one time homeostasis is disrupted, the NLRP3 inflammasome volition sense danger signals and actuate accordingly. Potassium efflux is a well-known activator of the NLRP3 inflammasome (46, 47). HCV infection induces potassium efflux in macrophages, thus leading to the maturation of pro-IL-1β (21).
Viroporins are small-scale, highly hydrophobic proteins derived from viruses, which interact with membranes to change the host cell's permeability to ions or other pocket-size molecules (48). Several viroporins are observed to localize to the Golgi apparatus and other cytoplasmic structures during viral infection (49–51). Examples include 2B proteins from EMCV, poliovirus, enterovirus 71 (EV71), and man rhinoviruses (HRV), the envelope (E) protein of astringent acute respiratory syndrome coronavirus (SARS-CoV), as well as influenza virus M2 poly peptide. These viroporins activate the NLRP3 inflammasome past inducing different ionic fluxes. Other viral proteins, such as non-structural 2B proteins from EMCV, HRV, poliovirus and EV71, as well as N protein from SARS-CoV, cause the flux of calcium from intracellular storages to the cytosol, which is indispensable for NLRP3 activation (49–51). The HCV core protein regulates intracellular calcium flux through a phospholipase C-dependent process, instead of directly changing the membrane permeability (52). HRV infection also induces the co-localization of NLRP3 and NLRC5, which sense calcium fluxes and assemble in a cooperative manner (50). Influenza virus M2, RSV minor hydrophobic (SH) protein, and SARS-CoV viroporin 3a change membrane permeability by forming a cation-selective ion channel. As a result, the ion channel permits the release of Na+/K+, rather than Ca2+, to induce the NLRP3 inflammasome activation (53–55). The disturbance of ionic concentrations leads to mitochondria damage and the product of ROS, potentiating NLRP3 inflammasome activation (55).
Mitochondria damage is too a crucial activator of the NLRP3 inflammasome. Similar to lysosomal or endosomal maturation, mitochondria impairment also induces the production of ROS to actuate the NLRP3 inflammasome (56, 57). The RIP1-RIP3 complex, assembled later on viral infection, induces activation of the GTPase DRP1. DRP1 and then translocates to the mitochondria to mediate its aberrant fission and damage (42). It has been reported that the dengue virus, VSV, SeV, too as poly(I:C), induce NLRP3 inflammasome activation and are all dependent on the RIP1-RIP3-DRP1 pathway (42, 58). Even so, reports argue that ROS, brought about by mitochondria damage, is not essential for activation of the NLRP3 inflammasome (59). Instead, mitochondrial membrane potential induced past influenza or EMCV is required for activation of the NLRP3 inflammasome. Under the appropriate mitochondrial membrane potential, NLRP3 will translocate to the mitochondria to combine with mitofusin 2, a mediator of mitochondrial fusion (59).
Accumulation of misfolded protein aggregates is an important activation signal of the NLRP3 inflammasome. A well-known example is that of Alzheimer'southward illness (Advert), which is characterized past the accumulation of amyloid-β peptide (sixty). The ORF 8b of SARS-CoV forms intracellular aggregates through the valine residue at position 77 (61). This circuitous acts as the danger signal to induce endoplasmic reticulum stress and lysosomal damage, resulting in NLRP3 inflammasome activation.
Viral infection alters the plasma membrane integrity and ionic efflux, which could atomic number 82 to programmed jail cell death and induce the secondary activation of NLRP3 inflammasome. The process of viral replication causes lytic jail cell death and subsequent potassium efflux, which provides the 2nd bespeak for NLRP3 inflammasome activation (62). The PB1-F2 poly peptide from IAV induces oxidative stress and the alteration of mitochondrial calcium, leading to apoptosis and NLRP3 inflammasome activation (63).
Viral Evasion Strategies Targeting The NLRP3 Inflammasome
Optimal activation of host immunity is crucial for the elimination of invading viruses. However, viruses have evolved strategies to evade immune responses by limiting the activation of the NLRP3 inflammasome. Some viruses have been reported to suppress NLRP3 inflammasome activation to circumvent innate immunity and facilitate viral replication (Effigy 2).
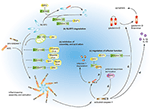
Figure 2. Viral allowed evasion strategies past targeting the NLRP3 inflammasome. (a) Influenza virus NS1 protein, measles virus, SeV, and Nipah virus V proteins prevent NRLP3 inflammasome associates. PB1-F2 of IAV and miR-BART15 of EBV inhibit NLRP3 inflammasome activation. (b) EV71 proteases 2A and 3C and HPIV C poly peptide induce NLRP3 protein degradation. (c) EV71 protease 3C and ZIKV NS1 protein modulate the effector function of the NLRP3 inflammasome by targeting GSDMD and caspase-1, respectively.
Viruses can inhibit both the assembly and the activation of the NLRP3 inflammasome through direct or indirect interactions. Measles virus and paramyxovirus (such every bit SeV and Nipah virus) Five poly peptide, and influenza virus NS1 protein inhibit NLRP3 inflammasome activation by interacting with NLRP3, decreasing the secretion of IL-1β accordingly (64–66). The interaction betwixt viral proteins and NLRP3 prevents the self-oligomerization of NLRP3 equally well every bit the recruitment of ASC, resulting in the cake of NLRP3-dependent ASC oligomerization and subsequent inflammasome activation (66). As a regulator of NLRP3 expression, the myeloid-specific microRNA miR-223 downregulates NLRP3 inflammasome activation past bounden within the three′ untranslated region (UTR) of NLRP3 (67). Under these conditions, EBV miR-BART15 specifically targets the miR-223 binding site in the NLRP3 3′-UTR to inhibit NLRP3 activation (68). miR-BART15 can also be transported around non-infected cells through exomes secreted from infected B cells, thereby amplifying this immunosuppressive state. PB1-F2, a viral virulence protein encoded by most IAV strains, could be spliced into different lengths (63). Although PB1-F2 induces excessive NLRP3 inflammasome activation by inducing apoptosis, the mediation of phagolysosome acidification or formation of PB1-F2 aggregates (22, 63), it too could impair NLRP3 inflammasome activation through different machinery (69). PB1-F2 distributes into the mitochondrial inner membrane space via Tom 40 channels and this mitochondrial location of PB1-F2 attenuates the mitochondrial membrane potential and every bit a result, inhibits NLRP3 inflammasome activation (69). These conflicting results may be attributed to the different secondary structures of PB1-F2 spliceosomes (70).
NLRP3 ubiquitination and poly peptide degradation is a key regulatory mechanism for NLRP3 inflammasome activation (71, 72). A couple of viral proteins mediate NLRP3 degradation and thus suppress NLRP3 inflammasome activation. The EV71 proteases 2A and 3C directly cleave NLRP3 protein at residues G493-L494 or Q225-G226, respectively (73). HPIV blazon 3 C protein interacts with NLRP3 and promotes its ubiquitination, thereby mediating NLRP3 proteasomal deposition (31).
A couple of viruses can modulate the effector functions of the NLRP3 inflammasome. The EV71 protease 3C cuts the NLRP3 inflammasome activation effect cistron GSDMD, at 193–194 residues, instead of 275–276 residues by caspase-one (74). This aberrantly cleaved GSDMD product fails to induce cell pyroptosis of the infected cell and, every bit a result, promote viral replication and attains the objective of viral evasion. Some viruses tend to take the "retreat in order to advance" strategy to maintain their survival. The ZIKV infection is a public wellness emergency and host IFN-β-associated antiviral innate immunity is essential for the command of this viral infection. Zheng and colleagues discovered that ZIKV infection induces NLRP3 inflammasome activation and deliberately enhances its activation through the NS1 protein (75). NS1 recruits the host deubiquitinase, USP8, to carve the polyubiquitin chains from caspase-1 so every bit to inhibit the proteasomal degradation of caspase-1 and, subsequently, amplify the NLRP3 activation signal. Yet, although the inflammatory response is strengthened by NS1, the large amount of caspase-ane turns out to the cleavage of cGAS, the critical component associated with the antiviral innate immune response. This interplay partly reflects the complex co-development between virus and host and may provide potential therapeutics in the future.
Decision And Perspective
Both the NLRP3 inflammasome activation and the subsequent inflammation play significant roles in defending confronting viral infections. However, abnormal NLRP3 inflammasome activation or chronic inflammation can also atomic number 82 to astringent pathological injury. Accordingly, activation of the NLRP3 inflammasome and its associated inflammation is a double-edged sword for host to defence force viral infection. Modulating the NLRP3 inflammasome action can evidence to be a promising strategy for the intervention of viral diseases. In a juvenile mouse model of IAV infection, MCC950, a specific inhibitor of the NLRP3 inflammasome, ameliorates astringent NLRP3 inflammasome-mediated lung injury without impairing viral clearance (76).
In this review, we focused on the activation of the NLRP3 inflammasome during viral infection, every bit well as the allowed evasion strategies of viruses. Yet, the mechanisms of NLRP3 inflammasome activation triggered by viral infection are far from fully elucidated and many questions still remain unanswered. Can NLRP3 sense viral components directly and, if and then, how? Are at that place any receptors that can coordinate with NLRP3 to recognize viruses? Can viral Deoxyribonucleic acid activate NLRP3 through the DNA sensors-RIP1-RIP3 pathway? A virus such every bit hepatitis B virus (HBV), could not actuate NLRP3 inflammasome by itself (77), so how does information technology escape the surveillance of the allowed system? Elucidating the mechanisms of NLRP3 inflammasome activation during viral infection volition help us to understand the pathogenesis of inflammation-associated diseases better and discover suitable therapeutic targets for viral diseases.
Author Contributions
CZ drafted the manuscript and figures. WZ supervised and edited the manuscript and figures.
Funding
This work was supported past grants from the National Natural Science Foundation of Cathay (81622030, 31870866, 81861130369, and 81901609), and National Cardinal Inquiry and Developmental Programme of People's republic of china (2017YFC1001100). WZ is a Newton Advanced Fellow awarded by the Academy of Medical Sciences.
Disharmonize of Interest
The authors declare that the research was conducted in the absenteeism of whatever commercial or financial relationships that could exist construed as a potential conflict of interest.
References
1. Tan XJ, Sun LJ, Chen JQ, Chen ZJ. Detection of microbial infections through innate allowed sensing of nucleic acids. Annu Rev Microbiol. (2018) 72:447–78. doi: x.1146/annurev-micro-102215-095605
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, et al. IL-1 beta signaling promotes CNS-intrinsic immune control of west nile virus infection. PLoS Pathog. (2012) eight:e1003039. doi: 10.1371/journal.ppat.1003039
CrossRef Total Text | Google Scholar
5. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, et al. The NLRP3 inflammasome mediates in vivo innate immunity to flu A virus through recognition of viral RNA. Immunity. (2009) 30:556–65. doi: 10.1016/j.immuni.2009.02.005
PubMed Abstract | CrossRef Full Text | Google Scholar
six. Swanson KV, Junkins RD, Kurkjian CJ, Holley-Guthrie East, Pendse AA, El Morabiti R, et al. A noncanonical function of cGAMP in inflammasome priming and activation. J Exp Med. (2017) 214:3611–26. doi: 10.1084/jem.20171749
PubMed Abstract | CrossRef Total Text | Google Scholar
8. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Jail cell. (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
PubMed Abstruse | CrossRef Full Text | Google Scholar
x. Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A crucial function of SGT1 and HSP90 in inflammasome action links mammalian and institute innate immune responses. Nat Immunol. (2007) 8:497–503. doi: 10.1038/ni1459
PubMed Abstract | CrossRef Full Text | Google Scholar
xi. Lo YH, Huang YW, Wu YH, Tsai CS, Lin YC, Mo ST, et al. Selective inhibition of the NLRP3 inflammasome by targeting to promyelocytic leukemia protein in mouse and human. Claret. (2013) 121:3185–94. doi: 10.1182/blood-2012-05-432104
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Shi JJ, Zhao Y, Wang K, Shi XY, Wang Y, Huang HW, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic prison cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
PubMed Abstract | CrossRef Full Text | Google Scholar
fifteen. He WT, Wan HQ, Hu LC, Chen PD, Wang 10, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-one beta secretion. Jail cell Res. (2015) 25:1285–98. doi: x.1038/cr.2015.139
CrossRef Full Text | Google Scholar
16. Niu JL, Wu SX, Chen MK, Xu K, Guo QH, Lu AL, et al. Hyperactivation of the NLRP3 inflammasome protects mice confronting flu A virus infection via IL-1 beta mediated neutrophil recruitment. Cytokine. (2019) 120:115–24. doi: ten.1016/j.cyto.2019.04.019
CrossRef Full Text | Google Scholar
17. Joosten LAB, Netea MG, Dinarello CA. Interleukin-1 beta in innate inflammation, autophagy and immunity. Semin Immunol. (2013) 25:416–24. doi: x.1016/j.smim.2013.10.018
CrossRef Full Text | Google Scholar
19. Coates BM, Staricha KL, Koch CM, Cheng Y, Shumaker DK, Budinger GRS, et al. Inflammatory monocytes bulldoze flu A virus-mediated lung injury in juvenile mice. J Immunol. (2018) 200:2391–404. doi: 10.4049/jimmunol.1701543
PubMed Abstract | CrossRef Total Text | Google Scholar
20. Saylor D, Dickens AM, Sacktor N, Haughey Northward, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder—pathogenesis and prospects for treatment. Nat Rev Neurol. (2016) 12:234–48. doi: 10.1038/nrneurol.2016.27
CrossRef Full Text | Google Scholar
21. Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, et al. IL-1β production through the NLRP3 inflammasome past hepatic macrophages links hepatitis C virus infection with liver inflammation and illness. PLoS Pathog. (2013) 9:e1003330. doi: x.1371/journal.ppat.1003330
PubMed Abstract | CrossRef Total Text | Google Scholar
22. McAuley JL, Tate Md, MacKenzie-Kludas CJ, Pinar A, Zeng Due west, Stutz A, et al. Activation of the NLRP3 inflammasome past IAV virulence protein PB1-F2 contributes to astringent pathophysiology and disease. PLoS Pathog. (2013) nine:e1003392. doi: 10.1371/journal.ppat.1003392
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Haque S, Lan X, Wen H, Lederman R, Chawla A, Attia One thousand, et al. HIV promotes NLRP3 inflammasome circuitous activation in murine HIV-associated nephropathy. Am J Pathol. (2016) 186:347–58. doi: 10.1016/j.ajpath.2015.10.002
PubMed Abstract | CrossRef Total Text | Google Scholar
24. Bauernfeind FG, Horvath One thousand, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
PubMed Abstract | CrossRef Total Text | Google Scholar
26. Poeck H, Bscheider M, Gross O, Finger K, Roth Due south, Rebsamen Thou, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin one beta production. Nat Immunol. (2010) 11:63–9. doi: ten.1038/ni.1824
PubMed Abstruse | CrossRef Full Text | Google Scholar
27. Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene Due east, von Messling V, et al. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. (2013) 9:e1003256. doi: 10.1371/journal.ppat.1003256
PubMed Abstract | CrossRef Total Text | Google Scholar
29. Segovia J, Sabbah A, Mgbemena V, Tsai SY, Chang TH, Berton MT, et al. TLR2/MyD88/NF-kappa B pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS One. (2012) 7:e29695. doi: x.1371/journal.pone.0029695
CrossRef Full Text | Google Scholar
30. Tsai SY, Segovia JA, Chang TH, Morris IR, Berton MT, Tessier PA, et al. DAMP molecule S100A9 acts as a molecular design to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog. (2014) ten:e1003848. doi: 10.1371/periodical.ppat.1003848
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Shil NK, Pokharel SM, Banerjee AK, Hoffman Thou, Bose Due south. Inflammasome antagonism by human being parainfluenza virus type 3 C poly peptide. J Virol. (2018) 92:e01776–17. doi: 10.1128/JVI.01776-17
PubMed Abstruse | CrossRef Total Text | Google Scholar
32. Wu JX, Sun LJ, Chen Ten, Du FH, Shi HP, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic Dna. Science. (2013) 339:826–thirty. doi: 10.1126/science.1229963
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Hernandez JC, Latz E, Urcuqui-Inchima S. HIV-1 induces the commencement signal to activate the NLRP3 inflammasome in monocyte-derived macrophages. Intervirology. (2014) 57:36–42. doi: 10.1159/000353902
PubMed Abstruse | CrossRef Full Text | Google Scholar
34. Mitoma H, Hanabuchi S, Kim T, Bao One thousand, Zhang Z, Sugimoto North, et al. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity. (2013) 39:123–35. doi: 10.1016/j.immuni.2013.07.001
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Chakrabarti A, Banerjee S, Franchi L, Loo YM, Gale M Jr, Nunez G, et al. RNase L activates the NLRP3 inflammasome during viral infections. Prison cell Host Microbe. (2015) 17:466–77. doi: 10.1016/j.chom.2015.02.010
PubMed Abstruse | CrossRef Full Text | Google Scholar
36. Li J, Hu Fifty, Liu Y, Huang L, Mu Y, Cai X, et al. DDX19A Senses viral RNA and mediates NLRP3-dependent inflammasome activation. J Immunol. (2015) 195:5732–49. doi: ten.4049/jimmunol.1501606
PubMed Abstruse | CrossRef Total Text | Google Scholar
37. Kuriakose T, Human SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of flu virus triggering the NLRP3 inflammasome and programmed cell expiry pathways. Sci Immunol. (2016) 1:aag2045. doi: 10.1126/sciimmunol.aag2045
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Rebsamen M, Heinz LX, Meylan East, Michallet MC, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. (2009) x:916–22. doi: 10.1038/embor.2009.109
PubMed Abstruse | CrossRef Full Text | Google Scholar
39. Yabal M, Muller N, Adler H, Knies N, Gross CJ, Damgaard RB, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. (2014) 7:1796–808. doi: 10.1016/j.celrep.2014.05.008
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Kesavardhana South, Kuriakose T, Guy CS, Samir P, Malireddi RKS, Mishra A, et al. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J Experi Med. (2017) 214:2217–29. doi: 10.1084/jem.20170550
PubMed Abstruse | CrossRef Full Text | Google Scholar
41. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, et al. Disquisitional role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. (2006) 281:36560–8. doi: ten.1074/jbc.M607594200
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Wang 10, Jiang Due west, Yan Y, Gong T, Han J, Tian Z, et al. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol. (2014) 15:1126–33. doi: 10.1038/ni.3015
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host Dna and triggers an innate immune response. Nature. (2008) 452:103–seven. doi: 10.1038/nature06664
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. (2008) 9:847–56. doi: 10.1038/ni.1631
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Barlan AU, Danthi P, Wiethoff CM. Lysosomal localization and mechanism of membrane penetration influence nonenveloped virus activation of the NLRP3 inflammasome. Virology. (2011) 412:306–xiv. doi: x.1016/j.virol.2011.01.019
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Mariathasan S, Weiss DS, Newton 1000, McBride J, O'Rourke M, Roose-Girma One thousand, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. (2006) 440:228–32. doi: 10.1038/nature04515
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Petrilli V, Papin South, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. (2007) 14:1583–ix. doi: x.1038/sj.cdd.4402195
PubMed Abstruse | CrossRef Full Text | Google Scholar
50. Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am J Respir Cell Mol Biol. (2013) 49:923–34. doi: 10.1165/rcmb.2013-0032OC
PubMed Abstruse | CrossRef Total Text | Google Scholar
51. Nieto-Torres JL, Verdia-Baguena C, Jimenez-Guardeno JM, Regla-Nava JA, Castano-Rodriguez C, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. (2015) 485:330–9. doi: 10.1016/j.virol.2015.08.010
PubMed Abstract | CrossRef Total Text | Google Scholar
52. Negash AA, Olson RM, Griffin Due south, Gale Yard Jr. Modulation of calcium signaling pathway past hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLoS Pathog. (2019) 15:e1007593. doi: 10.1371/journal.ppat.1007593
PubMed Abstruse | CrossRef Full Text | Google Scholar
54. Triantafilou Yard, Kar Southward, Vakakis Due east, Kotecha S, Triantafilou M. Human respiratory syncytial virus viroporin SH: a viral recognition pathway used by the host to indicate inflammasome activation. Thorax. (2013) 68:66–75. doi: 10.1136/thoraxjnl-2012-202182
PubMed Abstruse | CrossRef Full Text | Google Scholar
55. Chen IY, Moriyama K, Chang MF, Ichinohe T. Severe Acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. (2019) 10:l. doi: 10.3389/fmicb.2019.00050
PubMed Abstract | CrossRef Full Text | Google Scholar
57. Rawat P, Teodorof-Diedrich C, Spector SA. Human immunodeficiency virus type-1 single-stranded RNA activates the NLRP3 inflammasome and impairs autophagic clearance of damaged mitochondria in man microglia. Glia. (2019) 67:802–24. doi: 10.1002/glia.23568
PubMed Abstruse | CrossRef Full Text | Google Scholar
58. Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. (2013) 122:3405–xiv. doi: 10.1182/claret-2013-05-504449
PubMed Abstract | CrossRef Full Text | Google Scholar
59. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin ii is required for NLRP3 inflammasome activation later RNA virus infection. Proc Natl Acad Sci U.s.. (2013) 110:17963–8. doi: 10.1073/pnas.1312571110
PubMed Abstract | CrossRef Full Text | Google Scholar
sixty. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. (2008) 9:857–65. doi: 10.1038/ni.1636
PubMed Abstruse | CrossRef Total Text | Google Scholar
61. Shi CS, Nabar NR, Huang NN, Kehrl JH. SARS-Coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Jail cell Expiry Discov. (2019) five:101. doi: x.1038/s41420-019-0181-seven
PubMed Abstract | CrossRef Full Text | Google Scholar
62. da Costa LS, Outlioua A, Anginot A, Akarid K, Arnoult D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Prison cell Death Dis. (2019) 10:346. doi: 10.1038/s41419-019-1579-0
PubMed Abstract | CrossRef Full Text | Google Scholar
63. Chen WS, Calvo PA, Malide D, Gibbs J, Schubert U, Bacik I, et al. A novel influenza A virus mitochondrial protein that induces cell expiry. Nat Med. (2001) 7:1306–12. doi: x.1038/nm1201-1306
PubMed Abstract | CrossRef Total Text | Google Scholar
64. Komune North, Ichinohe T, Ito Thousand, Yanagi Y. Measles Virus V Protein inhibits NLRP3 inflammasome-mediated interleukin-ane beta secretion. J Virol. (2011) 85:13019–26. doi: x.1128/JVI.05942-11
CrossRef Total Text | Google Scholar
65. Moriyama 1000, Chen IY, Kawaguchi A, Koshiba T, Nagata Thou, Takeyama H, et al. The RNA- and TRIM25-binding domains of influenza virus NS1 poly peptide are essential for suppression of NLRP3 inflammasome-mediated interleukin-1 beta secretion. J Virol. (2016) 90:4105–fourteen. doi: 10.1128/JVI.00120-xvi
CrossRef Full Text | Google Scholar
66. Komatsu T, Tanaka Y, Kitagawa Y, Koide North, Naiki Y, Morita N, et al. Sendai virus V protein inhibits the secretion of interleukin-one beta by preventing NLRP3 inflammasomeassembly. J Virol. (2018) 92:e00842–18. doi: ten.1128/JVI.00842-18
CrossRef Total Text | Google Scholar
67. Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung Five. NLRP3 Inflammasome activeness is negatively controlled by miR-223. J Immunol. (2012) 189:4175–81. doi: ten.4049/jimmunol.1201516
PubMed Abstract | CrossRef Full Text | Google Scholar
68. Haneklaus M, Gerlic Thousand, Kurowska-Stolarska M, Rainey AA, Pich D, McInnes IB, et al. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1 beta production. J Immunol. (2012) 189:3795–9. doi: ten.4049/jimmunol.1200312
CrossRef Full Text | Google Scholar
69. Yoshizumi T, Ichinohe T, Sasaki O, Otera H, Kawabata SI, Mihara K, et al. Flu A virus poly peptide PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate amnesty. Nat Commun. (2014) 5:4713. doi: 10.1038/ncomms5713
PubMed Abstract | CrossRef Full Text | Google Scholar
70. Solbak SM, Sharma A, Bruns G, Roder R, Mitzner D, Hahn F, et al. Flu A virus protein PB1-F2 from different strains shows distinct structural signatures. Biochim Biophys Acta. (2013) 1834:568–82. doi: ten.1016/j.bbapap.2012.11.009
PubMed Abstract | CrossRef Full Text | Google Scholar
72. Vocal H, Liu BY, Huai WW, Yu ZX, Wang WW, Zhao J, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun. (2016) vii:13727. doi: 10.1038/ncomms13727
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Wang HB, Lei XB, Xiao X, Yang CF, Lu WL, Huang Z, et al. Reciprocal regulation between enterovirus 71 and the NLRP3 inflammasome. Cell Rep. (2015) 12:42–viii. doi: 10.1016/j.celrep.2015.05.047
PubMed Abstruse | CrossRef Total Text | Google Scholar
75. Zheng YY, Liu QX, Wu YX, Ma L, Zhang ZZ, Liu T, et al. Zika virus elicits inflammation to evade antiviral response past cleaving cGAS via NS1-caspase-i axis. Embo J. (2018) 37:e99347. doi: ten.15252/embj.201899347
PubMed Abstract | CrossRef Full Text | Google Scholar
76. Coates BM, Staricha KL, Ravindran N, Koch CM, Cheng Y, Davis JM, et al. Inhibition of the NOD-Like receptor protein three inflammasome Is Protective In Juvenile Influenza A virus infection. Front end Immunol. (2017) eight:782. doi: x.3389/fimmu.2017.00782
PubMed Abstract | CrossRef Total Text | Google Scholar
77. Yu 10, Lan PX, Hou XB, Han QJ, Lu N, Li T, et al. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1 beta production via suppressing the NF-kappa B pathway and ROS product. J Hepatol. (2017) 66:693–702. doi: x.1016/j.jhep.2016.12.018
CrossRef Full Text | Google Scholar
Source: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00211/full
0 Response to "Inflammasomes Mechanism of Assembly Regulation and Signaling Broz and Dixit Nat Imm Review"
Post a Comment